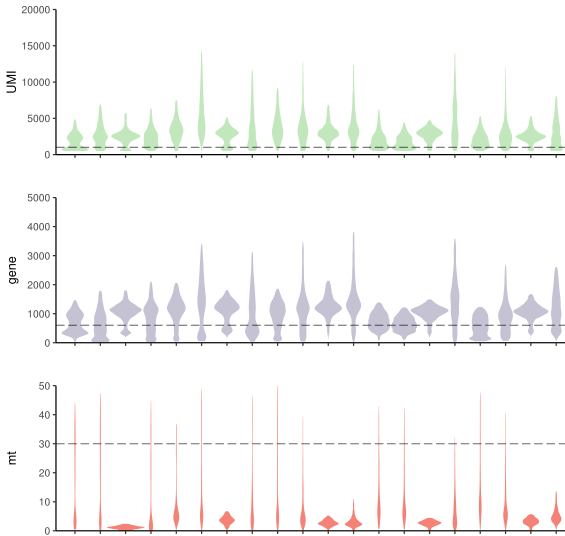
library(cowplot)
test.seu$patient=str_replace(test.seu$orig.ident,"_.*$","")
p1 <- DimPlot(test.seu, reduction = "tsne", group.by = "patient", pt.size=0.5)
p2 <- DimPlot(test.seu, reduction = "tsne", group.by = "ident", pt.size=0.5, label = TRUE,repel = TRUE) #后面两个参数用来添加文本标签
p3 <- DimPlot(test.seu, reduction = "umap", group.by = "patient", pt.size=0.5)
p4 <- DimPlot(test.seu, reduction = "umap", group.by = "ident", pt.size=0.5, label = TRUE,repel = TRUE)
fig_tsne <- plot_grid(p1, p2, labels = c('patient','ident'),align = "v",ncol = 2)
ggsave(filename = "tsne.pdf", plot = fig_tsne, device = 'pdf', width = 30, height = 15, units = 'cm')
fig_umap <- plot_grid(p3, p4, labels = c('patient','ident'),align = "v",ncol = 2)
ggsave(filename = "umap.pdf", plot = fig_umap, device = 'pdf', width = 30, height = 15, units = 'cm')
ident表示每个细胞的标签,聚类之后就是聚类的结果,在一些特定场景可以更换。
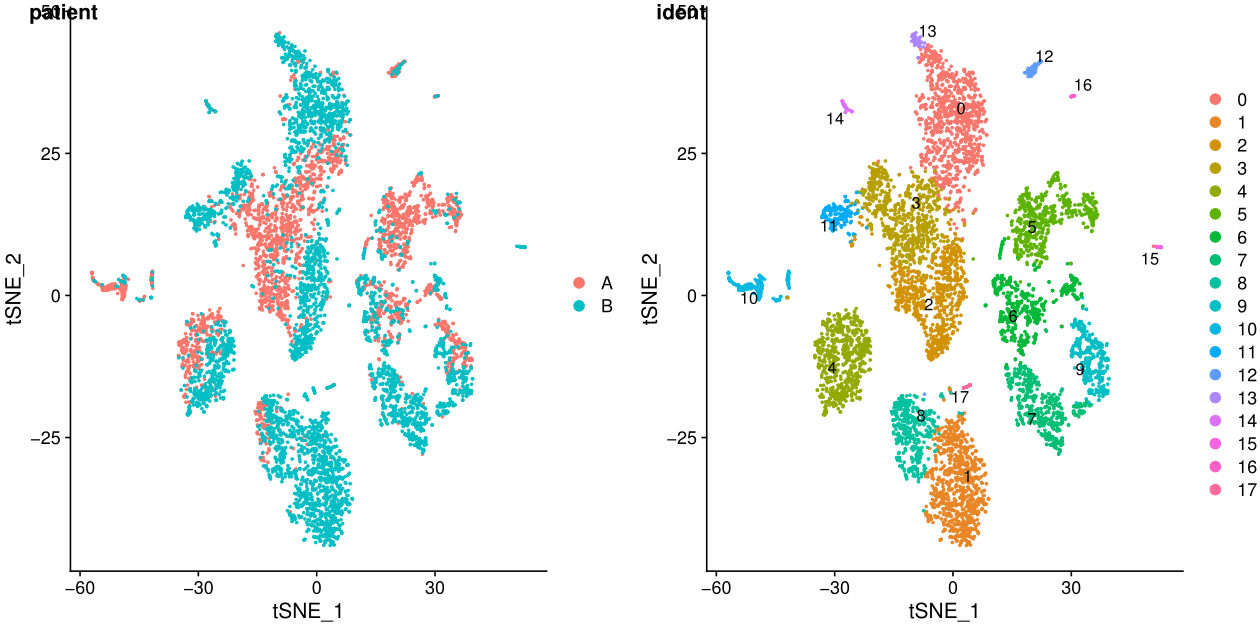
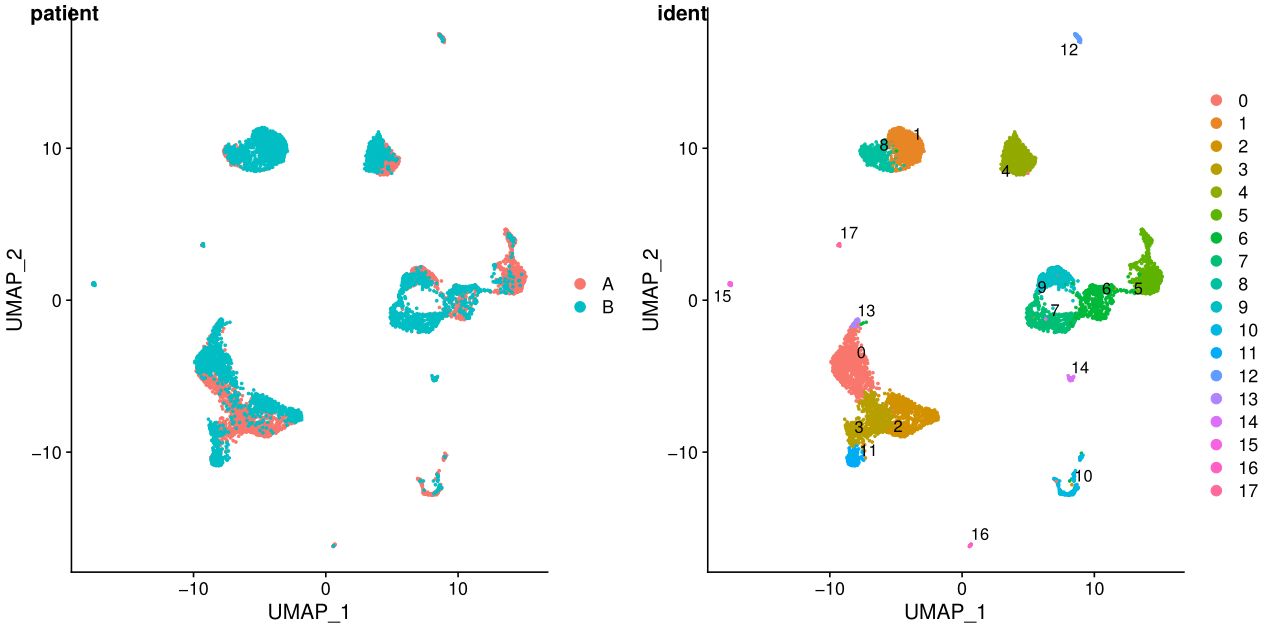
在umap图中,cluster之间的距离更明显
从上面的图可以看出不同样本其实是有批次效应的,下一讲我会介绍两种去批次效应的方法。
因水平有限,有错误的地方,欢迎批评指正!